我们 DNASTAR 有时会收到有关使用 PacBio HiFi 测序进行 DNA 组装的问题。我决定写一篇关于这个主题的博客文章是合适的。A 部分采用问答格式,以便轻松找到您感兴趣的信息。B 部分是如何在 Lasergene 中设置和分析 PacBio HiFi 组件的分步演示。
A 部分:PacBio HiFi 问答
在发送 DNA 进行测序时,为什么要选择 PacBio HiFi 而不是其他长读长或短读长测序技术?
PacBio HiFi 测序是一种尖端的 DNA 测序技术,可提供高精度的长读长测序。
传统的“短读长”测序方法产生相对较短的 DNA 片段。另一方面,PacBio HiFi 生成的读取时间要长得多,通常有数万个碱基对的长度。这对于解决复杂的基因组区域至关重要,例如重复序列和结构变异。
HiFi 测序读取的长度可达 30kb,并拥有高准确率(大于 99.9%),可与 Sanger 测序相媲美,这对于可靠的变体检出和基因组组装至关重要,即使在短读长测序技术无法准确对齐的高同源性和重复性区域也是如此。此外,虽然短读长技术可用于准确检测 SNP 和小插入缺失,但 HiFi 测序可以准确检测更广泛的变异,包括结构变异、相变和甲基化模式。
同样,虽然短读长测序技术被广泛用于RNA-seq基因表达分析,但全长mRNA转录本的解析很困难,而HiFi测序可用于解析全长mRNA转录本并精确分类选择性剪接事件。
使用 HiFi 数据时遇到哪些常见陷阱或挑战,以及如何避免这些陷阱或挑战?
我不知道有任何特定于 HiFi 数据的问题,这些问题也不属于短读取数据的问题。这两种数据类型确实可能会生成大量数据,需要强大的数据存储和计算资源。
文库制备和测序的最佳实践是什么,以最大限度地提高 HiFi 读取质量和产量?
为了最大限度地提高每个 SMRT 细胞的 HiFi 产量,PacBio 建议将 gDNA 片段化为 15 kb – 18 kb 之间的大小分布模式,以进行人类全基因组测序。不建议将大小分布模式大于 20 kb 的文库用于 HiFi 测序。
HiFi技术的未来发展方向和潜在应用是什么?
未来将扩展的一种应用是临床宏基因组学。目前,临床诊断依赖于培养方法,这些培养方法可能无法检测到低丰度细菌的存在或难以在培养基上生长的细菌。相比之下,HiFi 宏基因组测序不依赖于在培养基上培养细菌的能力,并且可以提供具有成本效益且全面的临床样本微生物概况。
HiFi 数据在准确性、成本和吞吐量方面与其他长读技术相比如何?你什么时候会选择一个而不是另一个?
一般来说,与纳米孔测序相比,HiFi 测序更准确(>99.9%),但读取长度更短(高达 25Kb),成本更高。纳米孔读取长度可超过 1Mb,并且更具成本效益,具有高通量潜力。当需要准确性时,例如复杂基因组区域的分辨率或单倍型相位,HiFi 是最佳选择,而对于大型项目或从头组装来说,纳米孔可能是更好、更具成本效益的选择,其中最长的读取可以跨越难以解析的重复区域。
哪些 SeqMan NGen 工作流程通常使用 PacBio HiFi?例如,我可以将其用于转录组分析或宏基因组学,还是仅用于全基因组/外显子组组装?
SeqMan NGen 支持 PacBio HiFi 数据的从头和参考引导组装和对齐。在 Lasergene 18.0 中,支持从头基因组(微生物)组装以及参考引导的基因组和外显子组比对。
在 SeqMan NGen 中使用 HiFi 读取时,我可以对杂合变体进行分相吗?如果是这样,如何?我在哪里分析结果?
是的,SeqMan NGen 提供了一种新的、新颖的单倍型定相算法,可以检测阶段性变异。分析在 GenVision Pro 中完成,其中可以可视化阶段区域(块)及其包含的变体。单个相位序列读数也可以用不同的颜色可视化,用于识别相位区块区域内的杂合等位基因。
在 SeqMan NGen 中组装 HiFi 数据(例如内存、CPU、GPU、磁盘空间)的计算要求是什么?有基于云的选项吗?
SeqMan NGen 的当前版本 (v18.0) 使用 CPU、可用磁盘空间和内存 (RAM) 的组合来对齐随数据集大小而变化的 PacBio HiFi 数据。对于人类基因组大小的数据,使用 8+ 核心 CPU、32GB RAM 和专用 4TB 硬盘来处理临时文件可获得最佳性能。SeqMan NGen (v18.1) 的下一次更新将能够利用 GPU 处理,大大提高 HiFi 数据的组装速度,并消除 4TB 可用磁盘空间要求。还有基于云的选项,允许用户在本地设置 HiFi 组件,然后自动压缩数据,上传到云端并在亚马逊云硬件上组装(然后自动下载)。对于想要并行处理多个数据集的用户,或者没有足够的本地计算资源来进行大型汇编项目的用户来说,这是一个很好的选择。
如何管理和存储HiFi测序生成的大数据集?
你需要很大的空间。我的个人电脑是一台功能强大的 i7 HP 笔记本电脑,带有两个硬盘(2TB 和 4TB)。我还使用外部 4TB 硬盘驱动器来存储额外的大型数据集。
B 部分:Lasergene 中的组装设置和下游分析
PacBio HiFi 数据(以及所有其他长读长类型)可用于从头组装和变异分析/重测序组装。以下示例显示如何设置和分析黑腹果蝇(果蝇)数据集的参考引导装配。
在 SeqMan NGen 中设置和运行程序集
1) 启动 SeqMan NGen 并单击New Assembly
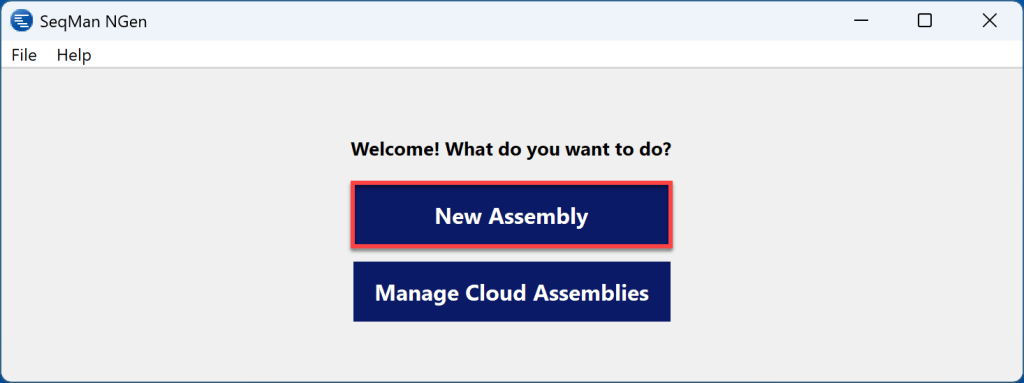
2) 在 SeqMan NGen Workflow 屏幕的Variant Analysis / Resequencing选项卡中,选择 PacBio / Nanopore Whole genome(全基因组)
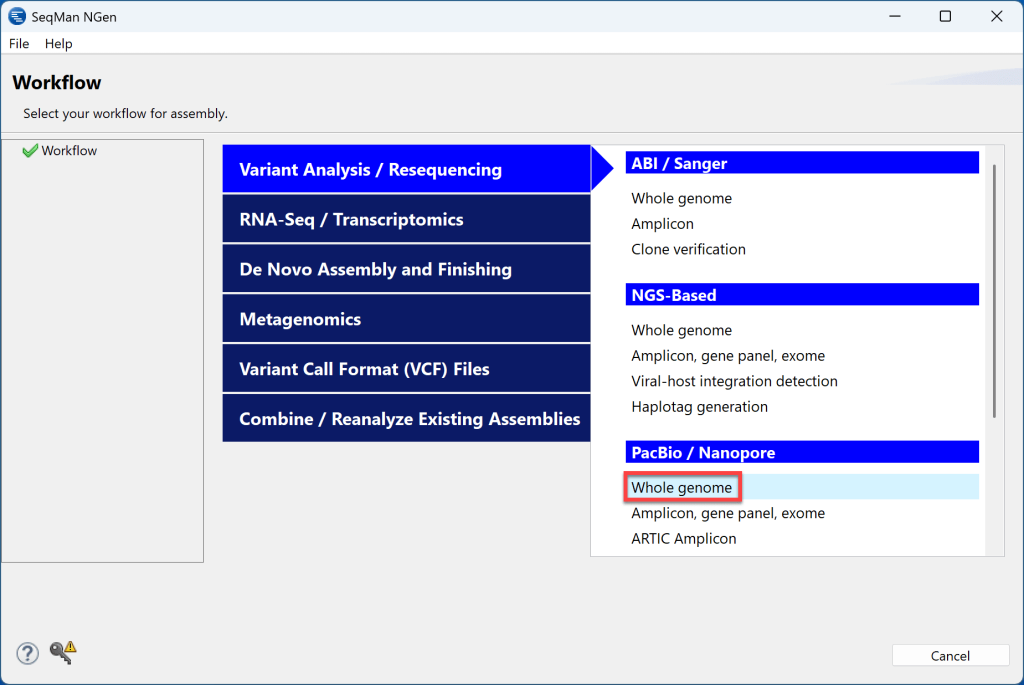
3) 在“参考序列”屏幕中,单击Download Genome Package以从 DNASTAR 中选择精选的模板包。
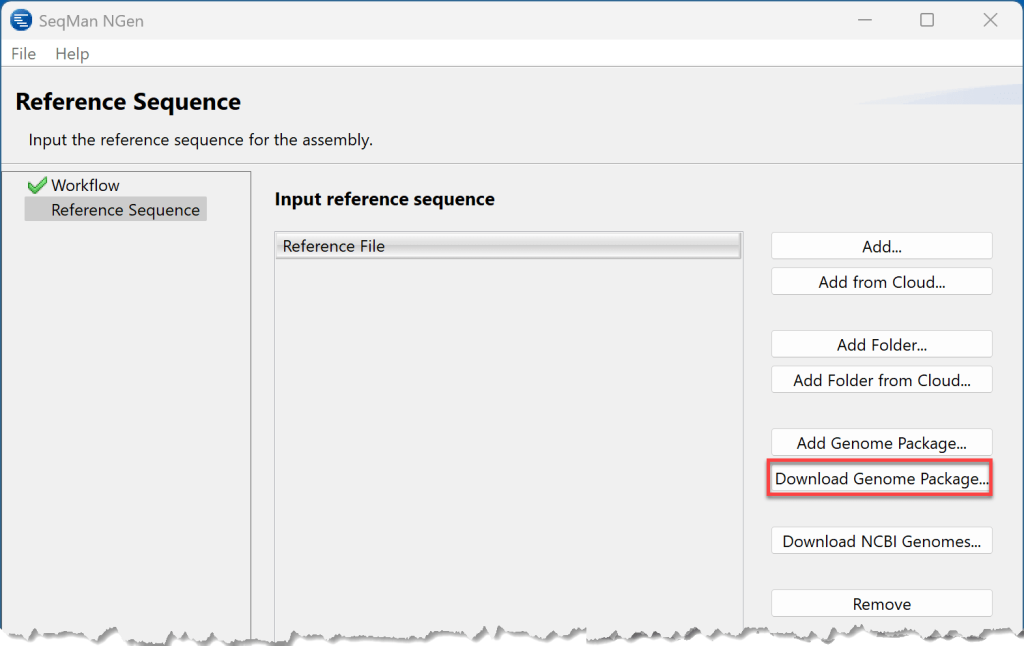
选择Drosophila melanogaster(黑腹果蝇),然后按Select。
然后按Next进入下一个屏幕。
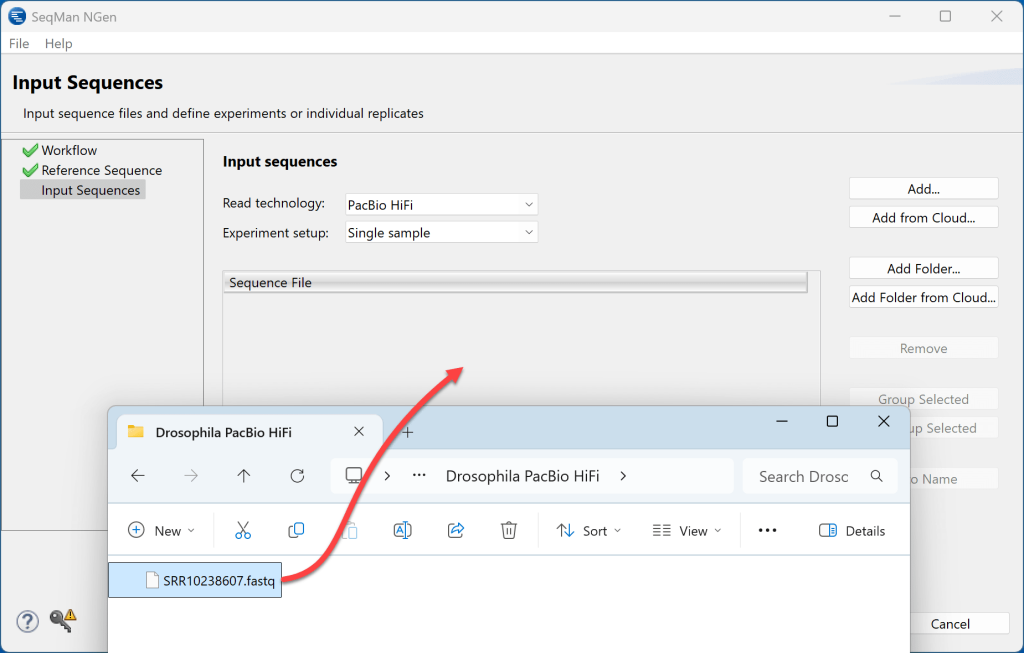
4) 在输入序列屏幕中,选择 PacBio HiFi 作为读取技术。使用Add按钮加载 PacBio HiFi 序列(此处为 50 MB .fastq 文件),或将其从文件资源管理器中拖放到 SeqMan NGen 向导屏幕上,如图所示。
5) 按两次Next进入Analysis Options屏幕。默认情况下,Detect SNPs and other small variants(检测SNP 和其他小变体)处于选中状态。由于生物体是二倍体,我们将采取选择二倍体的可选Diploid – Phased。这导致 SeqMan NGen 在组装过程中按等位基因分离变体,因此我们可以在分析过程中查看分阶段变体。我们不知道果蝇的性别,因此我们选择Unknown。
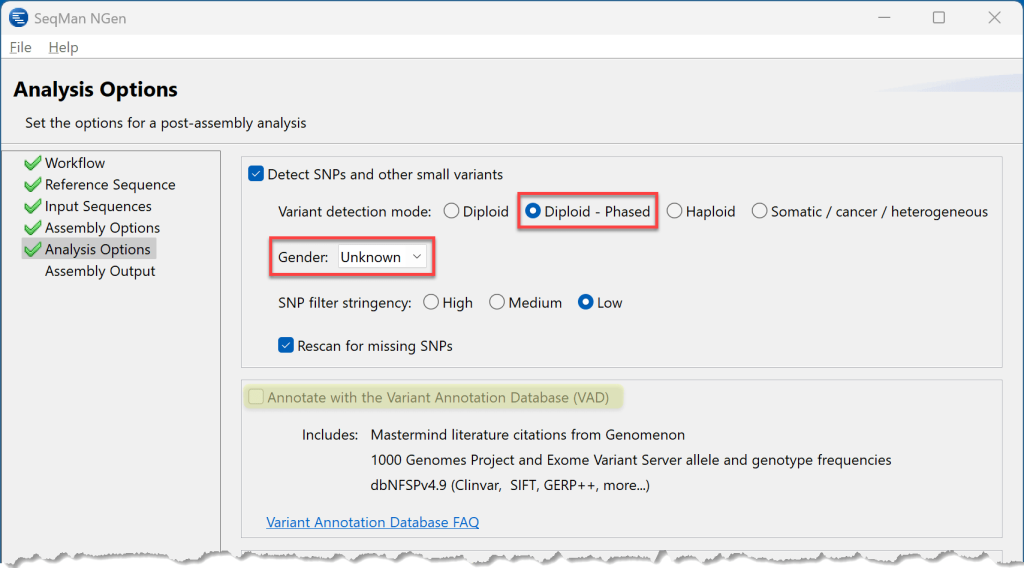
(顺便说一句,如果我们一直在处理人类样本,并希望自动为组装过程中发现的变体添加增强注释,我们会在步骤 3 中选择 Human build 37 或 38。然后,在此步骤中,我们将选中Annotate with the Variant Annotation Database旁边的框,上面以黄色突出显示。
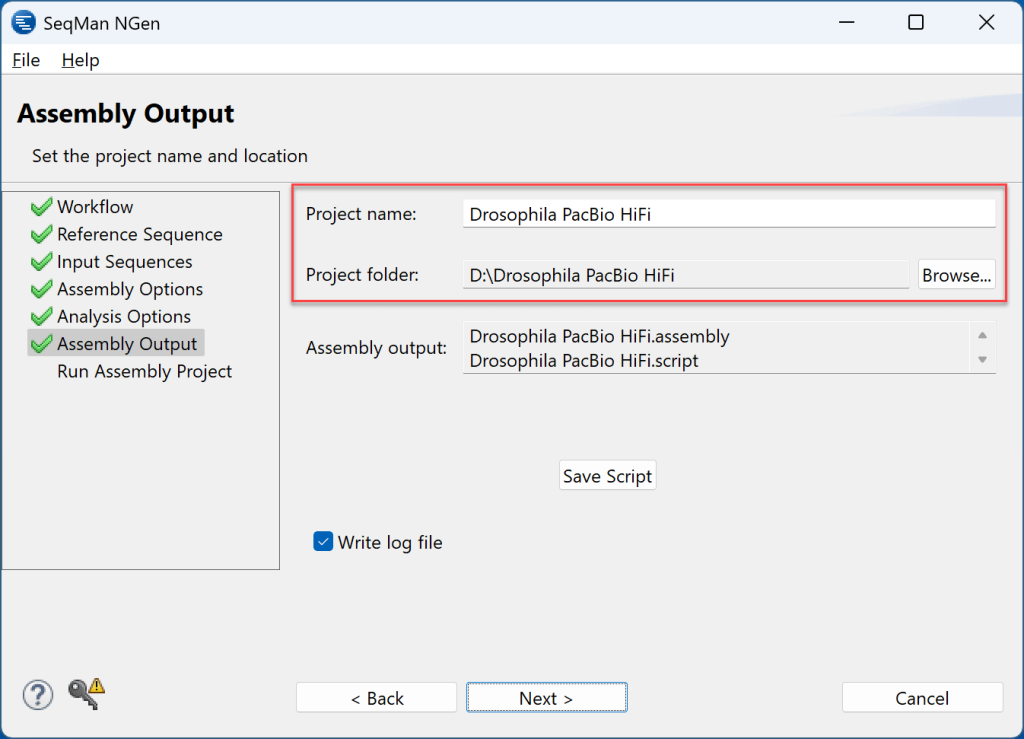
6) 单击Next进入装配输出屏幕。在那里,选择一个项目名称并分配要保存项目的文件夹。
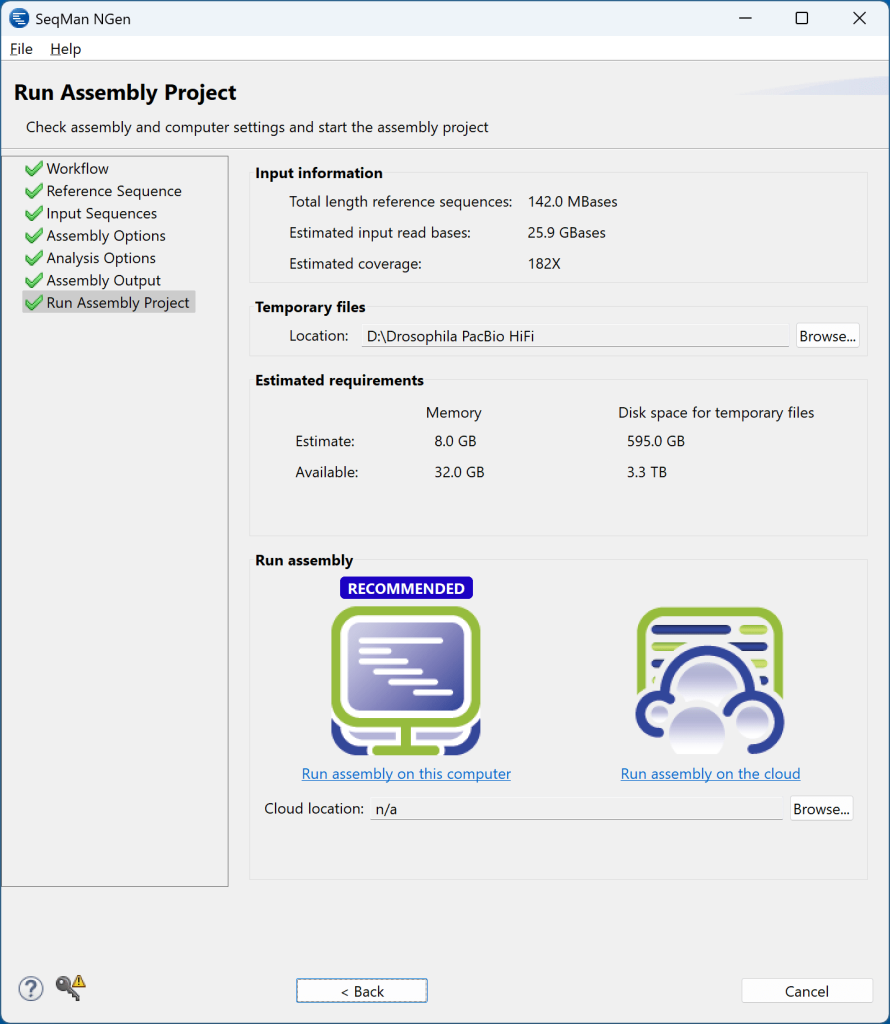
7) 单击Next 移至“运行装配项目”(Run Assembly Project) 屏幕。在这种情况下,SeqMan NGen 已经检查了我们计算机的可用内存,并建议在本地运行程序集。我们按链接 “Run assembly on this computer(在此计算机上运行程序集)” 启动程序集。
8) 装配完成后,短语“XNG done”将出现在装配日志的底部,并且右下角的Finish 按钮将变为活动状态。
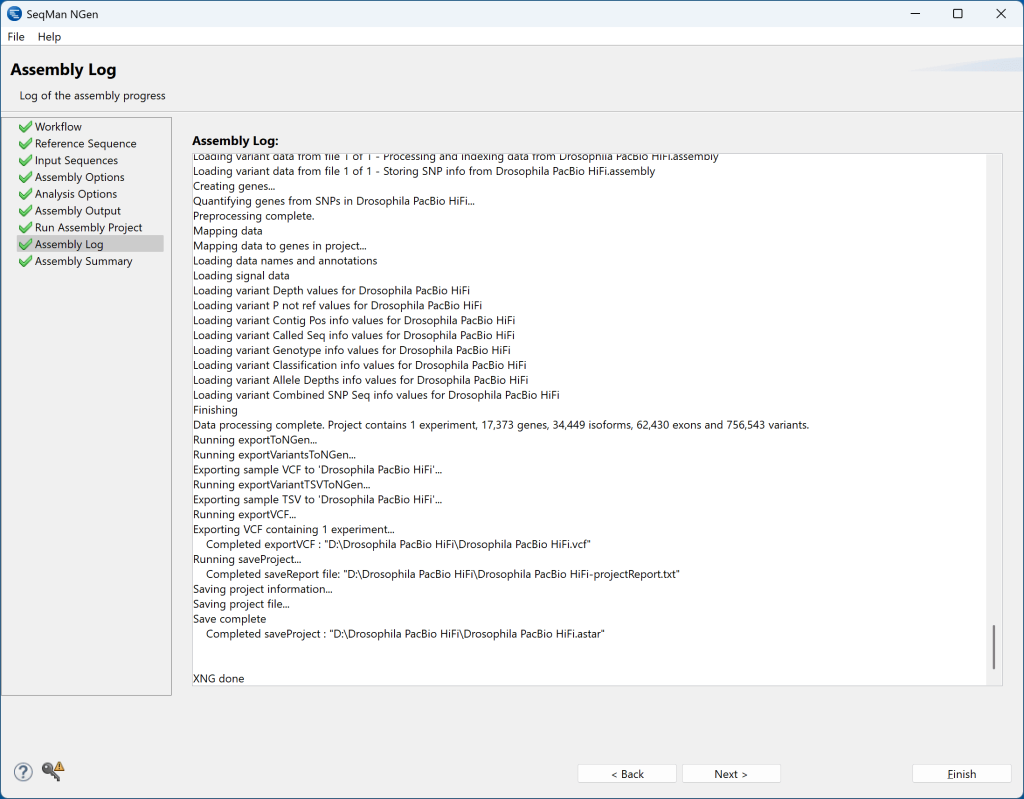
9) 按Next 进入Assembly Summary屏幕。
10) 要在 GenVision Pro 中打开装配,请单击Analyze and compare variants按钮。
在 GenVision Pro 中分析变体
在 Genome Pro 中,左侧的基因组视图将每个染色体显示为一排蓝色和绿色交替的相位块,而右侧的实验面板列出了染色体及其长度。
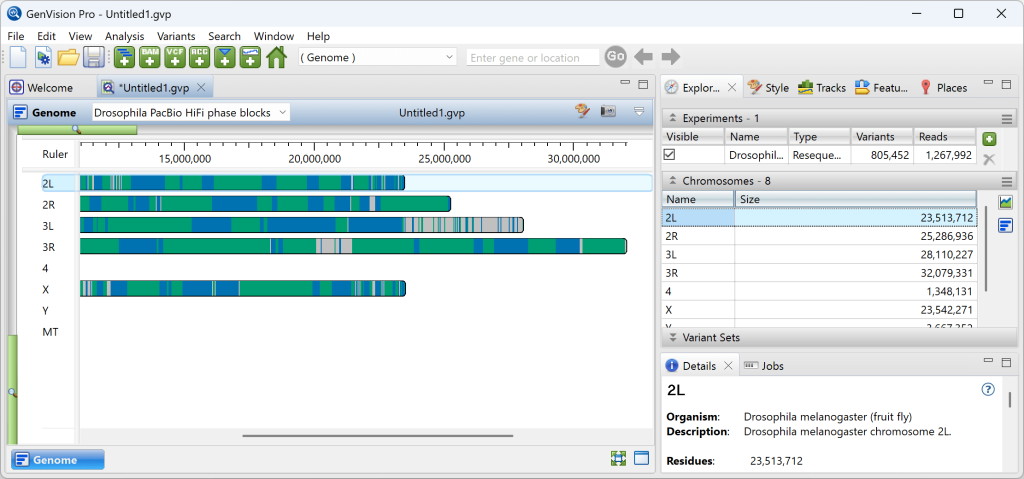
1) 要在Analysis 视图中更详细地查看染色体,请在任一位置双击其所在行。缩小视图后,顶层视图将显示特征和相位块。在下图中,您可以看到绿色相位的结束位置和蓝色相位的开始位置。对比色的垂直线表示这些位置的变异。
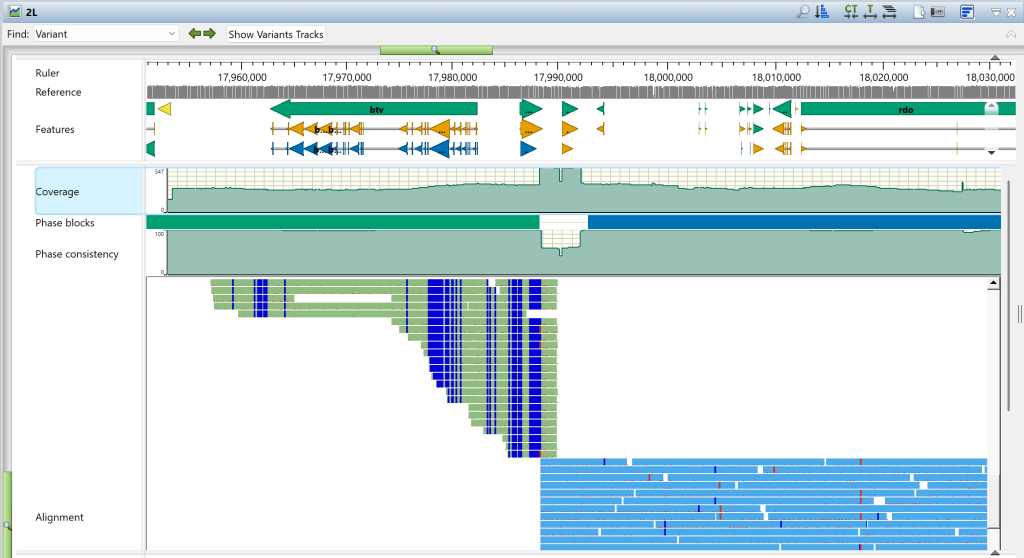
2)按下右上角的CT或T工具放大查看单个碱基和变异。下图中,在 Pde11 基因内,18558809 位有一个变异。参考行显示该位置有一个“T”。在比对轨道中,绿色显示的等位基因也有一个“T”,但蓝色显示的等位基因有一个变异“A”。下方的变异轨道以图形方式显示了相同的情况。请注意,变异轨道分为两个部分,一个等位基因中有变异(短竖线),另一个则没有。

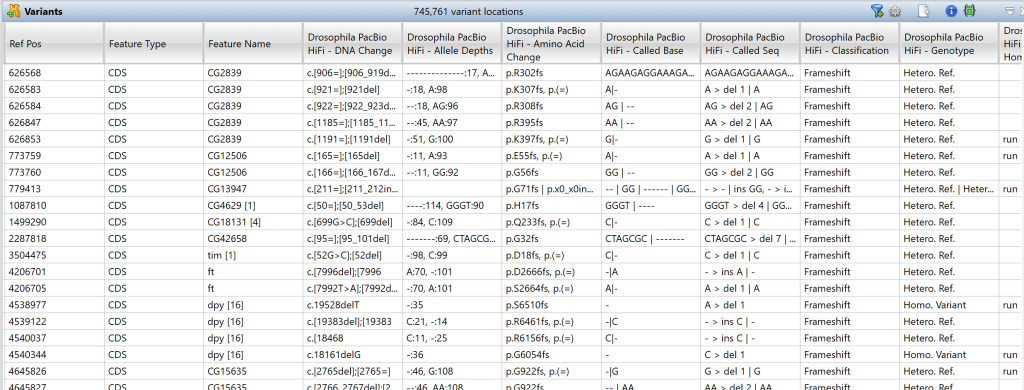
3) 要创建变体表,请单击Experiments 面板中的Show Variants Table 工具。
大量可自定义的数据列提供了每个变体的统计信息和其他有价值的信息。下方的变体表格仅显示了这些列的一部分。
结论
PacBio HiFi 的准确性和长读长能力正在改变基因组研究。Lasergene 的 SeqMan NGen 简化了此类数据的组装和分析,提供直观的设置界面、内置变异检出以及单倍型定相等功能。对于下游分析,GenVision Pro 提供多种视图和众多自定义选项,包括强大的变异过滤功能。
作者:DNASTAR 高级产品经理 Matt Keyser2025年4月14日 | Lasergene Genomics
作为 Lasergene 的高级产品经理,Matt Keyser 与 DNASTAR 的科学家、软件开发人员和支持人员携手合作,开发出既能满足研究人员当前需求,又能应对未来挑战和技术变革的序列分析软件。在 DNASTAR 工作的 20 年(以及未来)中,Matt 为众多客户提供了各种测序和分析项目的咨询服务,这让他对当今科学家面临的挑战有着独到的见解。